

McArdle disease is also referred to as glycogen disorder type V, this rare condition is an inborn metabolic abnormality that features a feature a deficiency or complete absence of an enzyme called glycogen phosphorylase (myophosphorylase).
McArdle disorder is a recessive inherited disorder that causes an inability to metabolize glycogen (the storage form of glucose in the body).
Persons with this condition present onsets of various symptoms mostly affecting the urinary and musculoskeletal systems.
McArdle disease was the first metabolic disease of the muscle (myopathy) to be recognized, it was brought to light by Dr. Brain McArdle while he was practicing in Guy’s Hospital London, England in 1951. He described the first case in a 30-year-old male who always experienced and complained of pain and weakness after exercise.
Dr. McArdle noticed the man’s cramps were electrically silent and his venous lactate levels failed to increase during an ischemic exercise.
The ischemic exercise is conducted by asking a patient to sneeze hard on a hand dynamometer for a specific period of time (usually a minute), while wearing a blood pressure cuff, which is placed on the upper arm and calibrated to 250 mmHg: blocking the flow of blood to the arm.
Upon exercise of any muscle, the lactate level of the blood is to increase, indicating the utilization of glucose by the muscle. But from Dr. McArdle’s deduction in his test, this process failed to occur.
Notably, this is the same event that occurs when a person suffers from a case of muscle poisoning by iodoacetate (a compound that blocks the conversion of glycogen into glucose and prevents the formation of lactate).
Dr. McArdle accurately concluded that the patient had a disorder of glycogen breakdown that primarily affected skeletal muscle. In 1959, the deficient enzyme associated with the condition was discovered by W.F.H.M Mommaerts et al.
McArdle disease usually presents itself in the second or third decade of life, but some cases have been reported at a relatively young age in neonates and infants presenting with hypotonia, generalized muscle weakness, and even respiratory failure. Patients of 40 years and above complain of weakness and wasting of muscles.
The exert statistical prevalence of McArdle disease was not particularly known as this rare inherited condition appeared to have a range from 1 in 50,000 to 1 in 200,000 in the United States.
A recent study analyzed gene frequency and next-generation sequencing data to report the true prevalence of the disease among populations.
The result from the study showed that the disease has a more common prevalence than previously stated, with a prevalence range of 1 in 5362 to 1 in 11,108.
The same study also employed an additional method to look into the two most common mutations causing the condition and recorded a prevalence of 1 in 42,355.
Causes of McArdle Disease
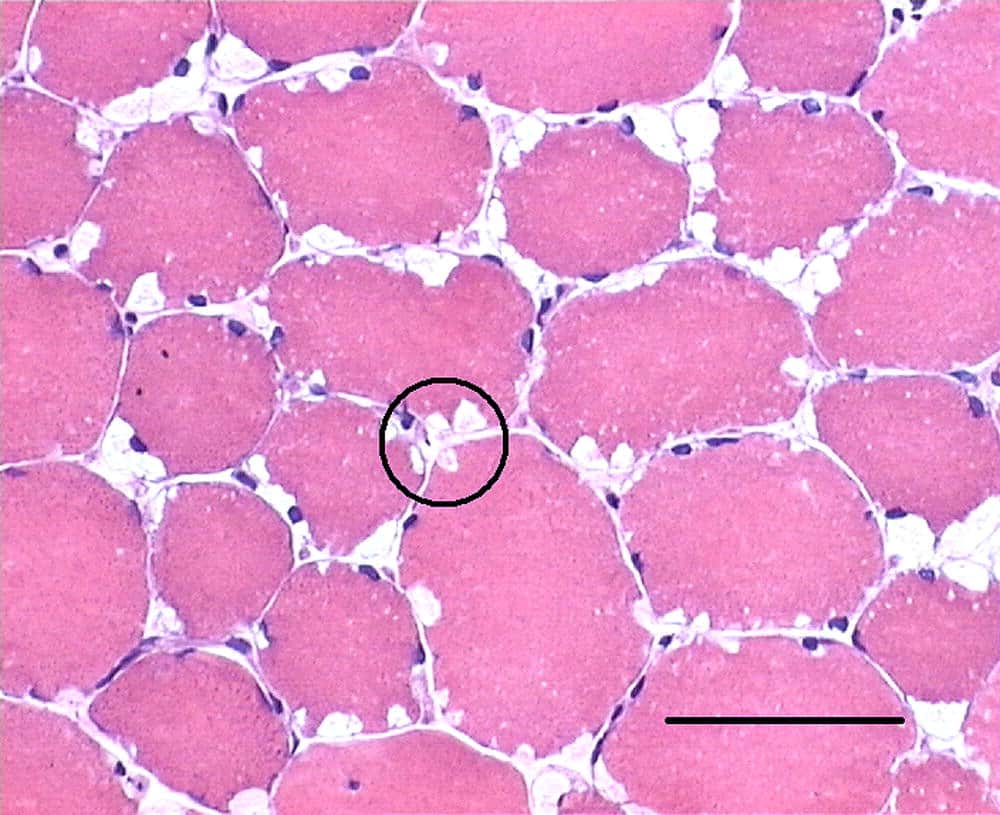
McArdle disease is an inherited condition of an autosomal recessive pattern, meaning both copies of the gene in each cell have mutations. Both parents of a person with an autosomal recessive condition each carry one copy of the mutated gene, but they do not show signs or present symptoms of the condition.
These mutations in found in the PYGM gene, responsible for glycogen phosphorylase enzyme (myophosphorylase) encoding. This enzyme function is key in the regulation of glucose metabolism in muscles.
Pathophysiologically, it detaches 1,4 glycosyl chains from glycogen and attaches inorganic phosphate to form glucose-1-phosphate; during glycogen breakdown, muscle cells generate glucose-1-phosphate in place of glucose, and due to the polar nature of the former molecule it disintegrates intracellularly.
PYGM gene mutations prevents myophosphorylase from breaking down glycogen effectively and as a result, muscle cells cannot utilize enough energy, so muscles become easily weary.
Reduced energy production in muscle cells leads to the major features of McArdle disease also referred to as glycogen storage disease type V (GSDV).
When mutated PGYM genes are inherited from both parents, it results to a full-blown McArdle disorder in the offspring.
Other terms used to refer to McArdle include:
- Gglycogen storage disease (GSD V)
- McArdle syndrome
- McArdle type glycogen storage disease
- McArdle’s disease
- PYGM deficiency
- Myophosphorylase deficiency
- Muscle phosphorylase deficiency
- Muscle glycogen phosphorylase deficiency
- Glycogenesis V
Clinical presentations and Symptoms of McArdle Disease
The earliest and most common symptoms of McArdle’s disease are typically present 10 seconds after beginning strenuous exercises, this is when the muscles rely on the conversion of glycogen to glucose to produce ATP (anaerobic period), the main energy soruce for muscle contration.
Symptoms may include:
- Fatigability
- Exercise intolerance
- Myalgia (muscular pain)
- Muscle contractures
- Stiffness
- Cramping (electrically silent contractions)
- Rhabdomyolysis (a breakdown of muscle tissue that releases a damaging protein into the blood)
- Myoglobinuria (presence of myoglobin in the urine)
- Premature Exhaustion: The inability of the skeletal muscles to metabolize glycogen into glucose with strenuous activity, leading to a sudden feeling of exhaustion or fatigue and an increase in heart rate. Patients complain of difficulty with any moderate activity such as walking, jogging and climbing steps.
- Muscle failure: This occurs under extreme stress when the muscle no longer can produce contraction regardless of the effort made, it leads to cramping which is very painful and results in muscle damage.
- Second wind: This clinical presentation is a phenomenon that has been observed in most patients with McArdle’s disease, it is characterized by a metabolic pathway switching from glycolytic to oxidative phosphorylation.
After 10 seconds into exercise, all of the readily available glucose in the system is depleted and the patient begins to fatigue. The patient experiences a moderate to severe pain from 10secs to 8mins of exercise due the muscles breaking down in order to compensate for the inability to break down glycogen.
After these few minutes, the second wind will kick in and the patient should be able to continue exercising for 45mins without pain and with an appropriate heart rate response, depending on the level of the exercise.
Second wind is not a systemic effect and must be achieved in each muscle group independently. This is not to relieve muscle failure symptoms for intense exercise but the body’s way of offering some relief for light to moderate exercises. It can be observed by monitoring a patient’s heart rate.
Diagnosis of McArdle Disease
Diagnosis of McArdle disease is done by laboratory tests, a muscle biopsy will reveal the absence of myophosphorylase in the muscle fibers. In other cases, acid-Schiff-stainedglycogen can be seen with microscopy.
The Ischemic Forearm Test is a valuable diagnostic tool for many metabolic conditions as it measures the concentration of lactic acid in the blood before and after a local exertion of a muscle group.
Another kind of test used for diagnosing McArdle’s disease is the Non-Ischemic Forearm Test; the test is done in the same manner as the ischemic forearm test but without the blood pressure cuff.
One of this method for this as recommended by Kazemi-Esfarjani et al. is the aerobic forearm test at 100% maximal voluntary handgrip (MVC), to be used as the routine diagnostic test for patients with suspected disorders of muscle carbohydrate metabolism.
Genetic sequencing of the PYGM gene (which codes for the muscle isoform of glycogen phosphorylase) may be done to determine the presence of gene mutations, to confirm the presence of McArdle disease.
This test is far less invasive than muscle biopsy. Urinalysis (urine test) is required only if rhabdomyolysis is suspected; urine volume, urine sediment, and myoglobin levels would be ascertained.
If rhabdomyolysis is suspected, serum myoglobin, creatine kinase, lactate dehydrogenase, electrolytes, and renal function will be checked.
Differential Diagnosis of McArdle Disease
Rhabdomyolysis is a biochemical syndrome and a clinical presentation of McArdle disease resulting from skeletal muscle injury that damages the structure of the muscle cell membranes sufficiently to allow the release of the muscle cell contents into the plasma. Rhabdomyolysis can exist independently or as a symptom of other conditions apart from McArdle disease.
Muscular Dystrophy is a group of more than 35 inherited conditions. They all cause muscle weakness and muscular integrity loss. Some forms of muscular dystrophy appear in infancy or childhood while others may not appear until middle age or later. The various types may vary in individual affected, muscle affected, and other symptoms are. All forms of muscular dystrophy grow worse as the person’s muscles get weaker; this may mean losing the ability walk. Weakness and muscle loss are some other symptoms shared by Muscular dystrophy.
Myositis is the inflammation of the muscles that moves the body. It may be caused by an injury, infection or an autoimmune disease. it exists are polymyositis which causes muscle weakness in the muscles closest to the trunk of the body and dermatomyositis which causes muscle weakness plus a skin rash. Myositis may be sometimes confused with McArdle disease because both conditions cause muscle weakness.
Another condition that may be mistaken for McArdle disease is Tarui’s Disease, a condition in which there is a deficiency of the phosphofructokinase enzyme; the enzyme needed to facilitate the breakdown of glucose into energy in muscle during exercise. Tarui’s disease is also a glycogen storage deficiency condition and so it has many similarities with McArdle disease.
Treatment of McArdle Disease
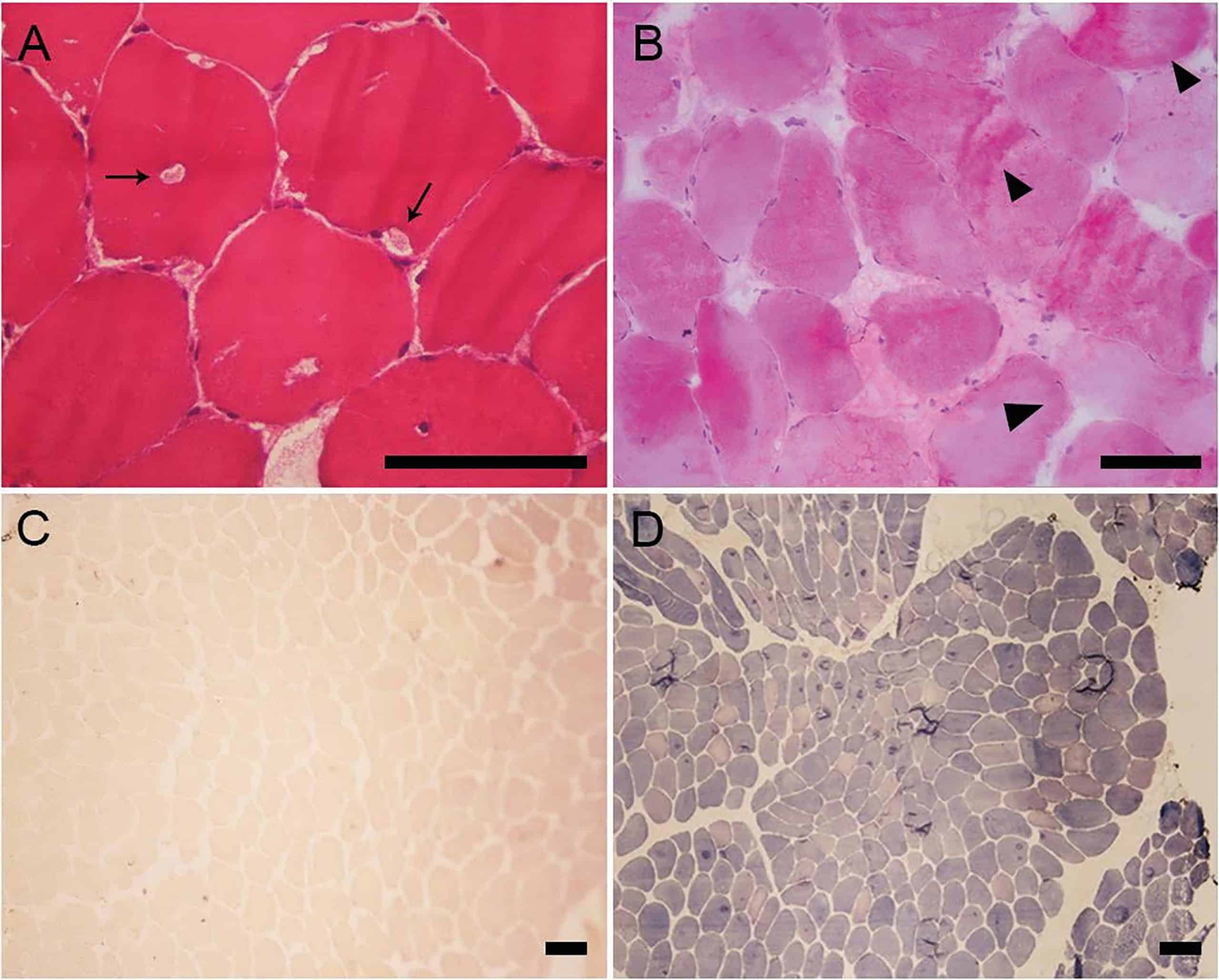
There is no specific treatment or management for persons with McArdle disease. Treatments trial usually revolves around avoiding lifestyle activities that exacerbate the symptoms.
Most persons adapt themselves by not engaging in strenuous exercises, but this may worsen the disease because serum CK (Creatinine Kinase) rises during the period of inactivity. It may also result in the inability of the muscles to utilize alternate fuels to overcome the block of glycogenolysis.
Certainly, dietary interventions which confer favourable effects include taking a sugeray meal before planning exercise such as oral sucrose treatment taken 30 minutes prior to exercise has been shown to help improve exercise tolerance including a lower heart rate and lower perceived levels of exertion compared with placebo. A diet rich in carbohydrates results in much better outcomes when compared to other nutrient-rich diet.
Other nutritional compounds that have been shown to be helpful for patients but could not yield convincing outcomes during actual experimental studies include branched-chain amino acids, depot glucagon, verapamil, vutaminb6, high dose D-ribose, dantrolene sodium and high-dose creatine ingestion.
Summary
McArdle disease is genetic condition discovered in 1951 by Dr. Brain McArdle in London. This condition among other disease is a glycogen storage deficiency, presenting itself mostly during the second and third decade of life. It can be diagnosed with some simple lab test, but there is no particular treatment.
The condition can only be managed by adopting a non-strenuous lifestyle and using nutritional supplements to compensate for glycogen deficiency before exercises or activities in order to compensate for glycogen deficiency.
Some other complication may arise in persons with McArdle disease when the course of the disease is altered with events of intensive exercise resulting in severe contractures and acute rhabdomyolysis.
This muscle breakdown causes the liberation of myoglobin (muscle protein) in blood and urine, and renal failure may consequently follow.
Sources;
- McArdle Disease; http://www.musculardystrophyuk.org/about-muscle-wasting-conditions/metabolic-myopathies/mcardle-disease-factsheet/
- McArdle Disease; http://www.mountsinai.org/health-library/diseases-conditions/mcardle-disease
- McArdle’s Disease; https://patient.info/doctor/mcardles-disease-glycogen-storage-disease-type-v







")



